







协和医科大学胡卓伟教授团队发现急性早幼粒白血病的重要发病机制。2017年5月发表在《Cancer Cell》杂志上。
研究中使用了汉恒生物病毒产品:TRIB3过表达腺病毒!
简介
- 首先介绍一下,急性早幼粒细胞白血病(APL)是一种血液系统恶性肿瘤,是(AML)的一种特殊类型,急性髓细胞性白血病是髓系造血干/祖细胞恶性疾病。以骨髓与外周血中原始和幼稚髓性细胞异常增生为主要特征,临床表现为贫血、出血、感染和发热、脏器浸润、代谢异常等,多数病例病情急重,预后凶险,如不及时治疗常可危及生命。
APL由含C末端视黄酸受体α(RARα)融合的早幼粒细胞白血病蛋白(PML)N末端的嵌合癌蛋白驱动。 一般而言,PML-RARa充当RARα和非RARα靶基因的转录阻遏物并减弱PML核体(PML-NB)的组装和功能,PML本身逐渐成为一种关键的肿瘤抑制因子。
p53是一种肿瘤抑制基因。在所有恶性肿瘤中,50%以上会出现该基因的突变。由这种基因编码的蛋白质是一种转录因子,其控制着细胞周期的启动。许多有关细胞健康的信号向p53蛋白发送。
已有研究表明,TRIB3表达在AML细胞中增强,但单独TRIB3过表达不驱动白血病疾病,并且关于TRIB3在AML发病机制和造血发育中的作用知之甚少。
他们最近发现TRIB3通过与自噬受体p62的相互作用以及干扰自噬和泛素蛋白酶体系统的降解功能来控制几种肿瘤的发生和发展是至关重要的。

此次胡卓伟教授团队于2017年5月在《Cancer Cell》杂志上发表了其团队的最新研究成果。在本研究中,他们发现了应激反应蛋白TRIB3的表达与APL患者的急性早幼粒细胞白血病进展和对治疗的抗性呈正相关。
通过与ATRA或As2O3合作破坏TRIB3 / PML-RARα相互作用的TRIB3表达或α-螺旋肽的遗传抑制通过促进PML-RARα降解产生针对APL的协同治疗功效。
这些结果揭示了应激传感器TRIB3在APL发病机制中的关键功能,并建议TRIB3,特别是TRIB3 / PML-RARα相互作用,作为治疗APL的潜在治疗靶标。
研究中使用了汉恒生物病毒产品:TRIB3过表达腺病毒!
结果
1、高表达TRIB3可以促进Pml-Rara驱动APL
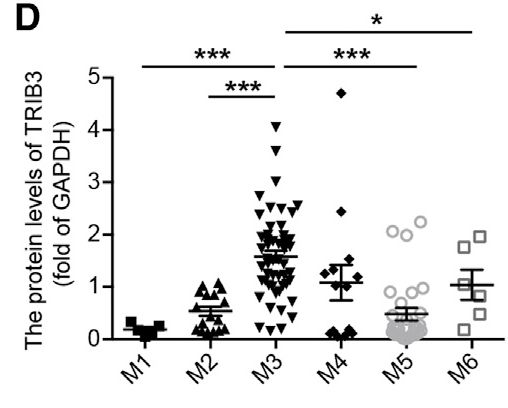
他们检测了来自健康供体的新诊断的AML患者与CD34 +正常细胞的CD34 +细胞的微阵列基因表达谱。在AML-M3(APL)患者中发现比具有M1,M2,M5和M6亚型AML的患者更高的TRIB3表达。
此外,TRIB3表达与APL患者BM中PML-RARα的表达正相关。

他们通过粒细胞/单核细胞集落形成单位评估了Trib3对BM中细胞自我更新的影响,表明Trib3通过支持Pml-Rara表达和APL细胞更新以及抑制PML-NB组装和分化来促进Pml-Rara驱动的APL。
2、降低TRIB3可以诱导APL细胞中p53介导的衰老
他们验证了TRIB3对照shRNA或表达TRIB3-shRNA1 / 2的NB4细胞的NOD-SCID小鼠中的作用。与表达TRIB3-shRNA1 / 2的小鼠相比,对照-shRNA小鼠表现出更高的白血病来源的生物发光性。
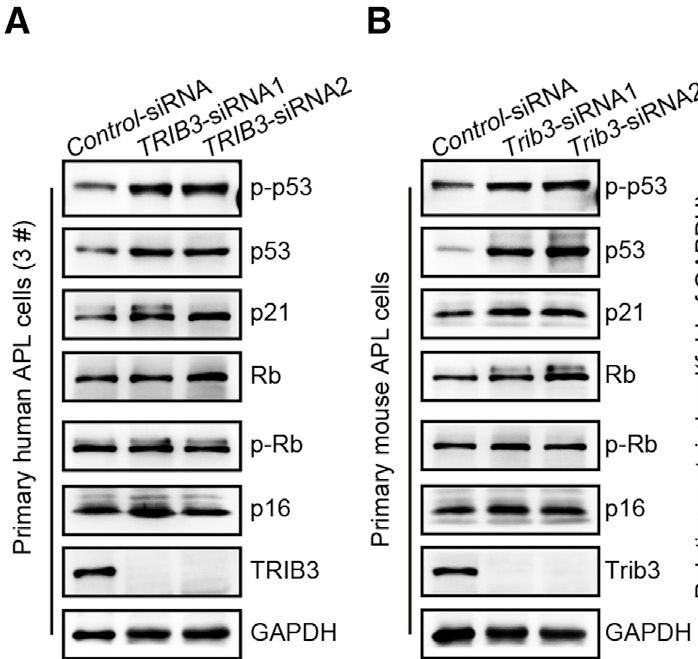
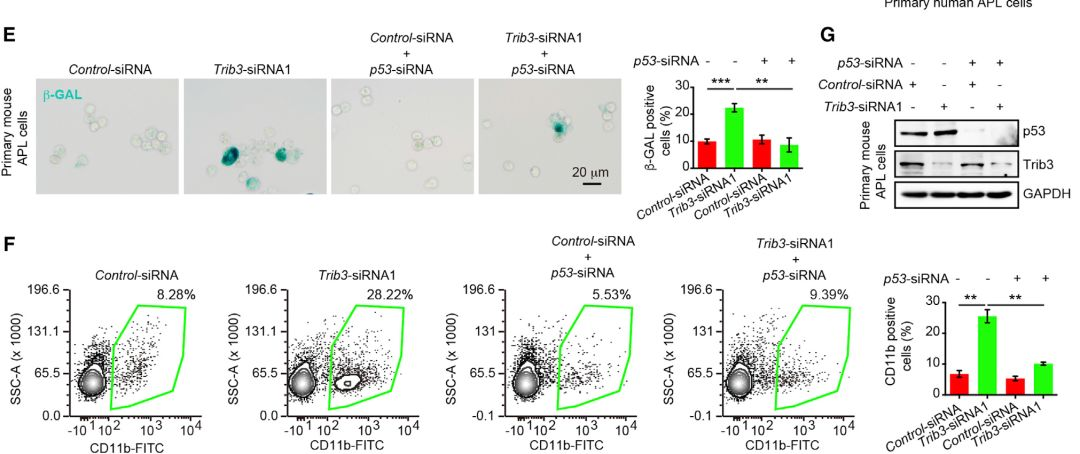
此外,在原代人APL细胞和原代小鼠APL细胞中,TRIB3消耗增加p53,p-p53和p21的表达,但不增加Rb,p-Rb和p16的表达。而且沉默p53逆转了原代小鼠APL细胞中Trib3耗尽诱导的衰老和分化。
表明TRIB3消耗通过在APL细胞中诱导p53介导的衰老和分化来抑制APL。
3、TRIB3抑制APL细胞中的PML-RARα降解
PML-NB完整性对于诱导p53依赖性衰老至关重要。因此,他们检查了TRIB3是否调控了PML-NB组装。在NB4细胞,原代小鼠APL细胞和原代人APL细胞中TRIB3的缺失增加了PML-NB数量。
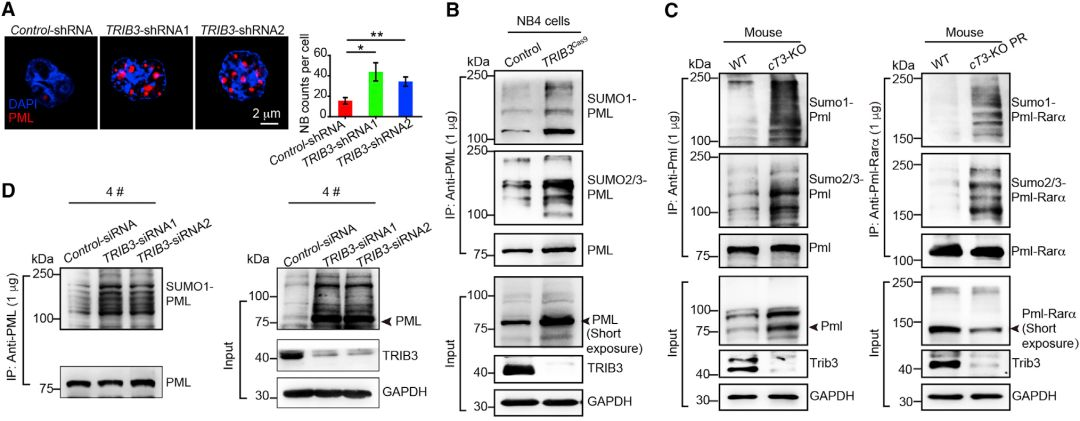
他们使用由纯化的E1(SAE1 / 2),E2(UBC9)和SUMO1组成的测定系统进一步评估PML总和的调节机制。他们发现E1激活了SUMO1,它在TRIB3存在下与UBC9缀合。此外,TRIB3抑制了SUMO1-缀合的UBC9与PML的结合,并抑制了PIAS1与PML的结合。
表明TRIB3通过阻碍E2或E3与PML的缔合并阻碍PIAS1从UBC9-SUMO复合物解离来抑制PML-RARα和PML的SUMO化。
高TRIB3表达抑制PML-RARα和PML的泛素化并延长其降解的半衰期。TRIB3的过度表达抑制As2O3诱导的PML-RARα转染的HEK293细胞中PML-RARα的SUMO化和泛素化。
这些数据表明TRIB3(使用了汉恒生物病毒产品TRIB3过表达腺病毒)通过抑制20S,11S和RNF4向PML-NBs的募集并抑制UPS活性来抑制PML-RARα和PML的泛素化和降解。
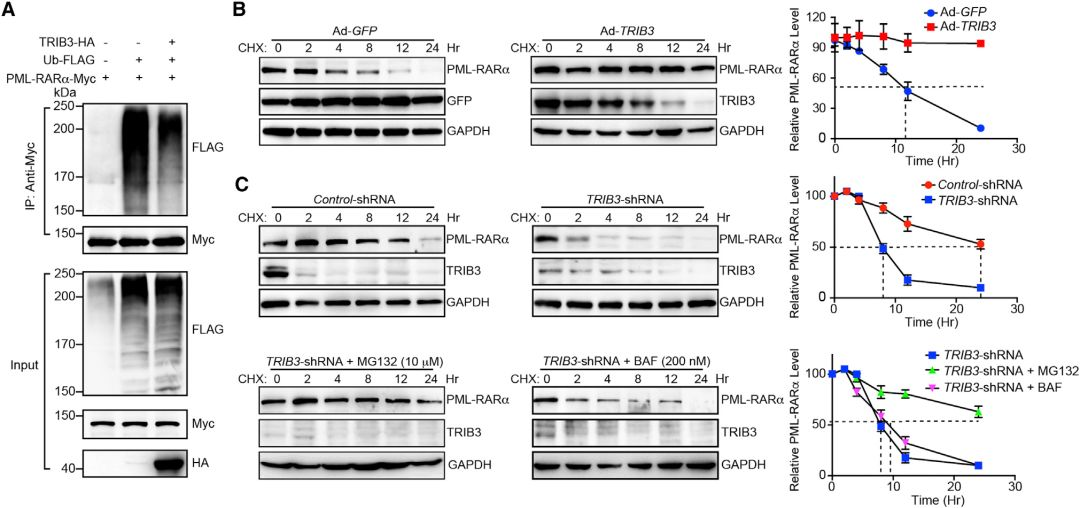
4、TRIB3与PML-RARα和PML相互作用抑制PML-RARα/ PML SUMO化
TRIB3通过蛋白质-蛋白质相互作用产生多种作用。PML-RARα在NB4细胞中与TRIB3共免疫沉淀,且TRIB3主要与嵌合PML-RARα的PML结构域相互作用。


使用PML结构域的缺失突变体,他们发现TRIB3与PML结构域的B-Box1、Ring-finger基序、全长蛋白质、PML的核定位信号结构域相互作用。


为了确定PML SUMO化基序的赖氨酸对于TRIB3 / PML相互作用是否必需,他们使用一系列PML赖氨酸突变体(K65R / K160R / K490R和K65A / K160A / K490A)来确定它们与TRIB3的关联。
他们发现K160和K490向精氨酸的突变降低了TRIB3 / PML相互作用,而K160和K490向丙氨酸的突变进一步降低了TRIB3 / PML相互作用。
5
与ATRA或As2O3协同干扰TRIB3 / PML-RARa相互作用根除APL
鉴于PML SUMO化的共有基序介导了PML / TRIB3相互作用,使用预测性I-TASSER服务器筛选了四种α-螺旋肽(S65,S160,S490和S497)的PML二级结构。
S160表现出对TRIB3的高亲和力。发现S160的残基11,12,13和14对于S160 / TRIB3结合是关键的。
他们通过将S160连接至先前鉴定的细胞穿透肽产生了融合肽Pep2-S160,并用Pep2-S160处理NB4细胞,抑制了TRIB3-PML相互作用,促进PML-RARα降解,挽救了由TRIB3表达引起的减少的PML SUMO化。且干扰了TRIB3 / PML-RARα相互作用并降低了白血病小鼠的BM和脾脏中的PML-RARα表达。

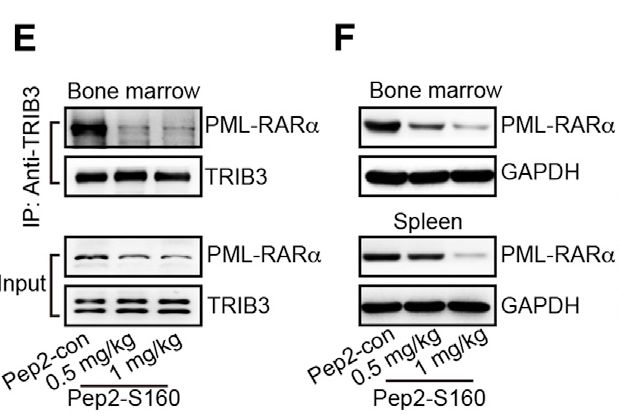
ATRA或As2O3治疗以时间依赖性方式增加TRIB3表达, Pep2-S160与ATRA或As2O3的组合不仅逆转了ATRA-或As2O3-增加的TRIB3表达,而且还增强了接种原代小鼠APL细胞的C57BL / 6小鼠中ATRA或As2O3的治疗效力以及接种NB4细胞的NOD-SCID小鼠。
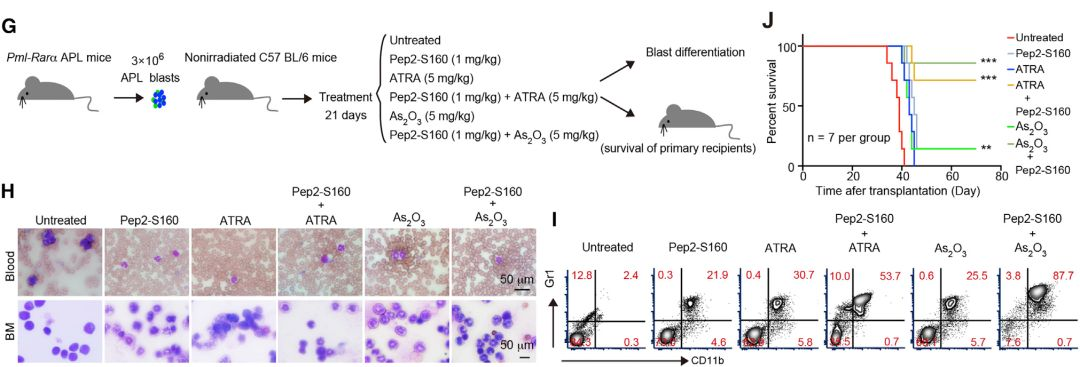
这些数据验证了TRIB3通过与PML和PML-RARα的相互作用在APL发病机制中起关键作用,并且扰乱这种相互作用加上ATRA或As2O3通过PML-RARα降解产生有效的抗白血病疗效。
总之,急性早幼粒细胞白血病由癌蛋白PML-RARα驱动,其对抗骨髓分化并促进APL起始细胞自我更新。

在此篇文章中,他们证明了TRIB3的表达与APL进展和治疗抵抗正相关。 升高的TRIB3表达通过与PML-RARα相互作用并抑制其SUMO化,泛素化和降解来促进APL,抑制了PML核体组装,p53介导的衰老和细胞分化,并支持细胞自我更新。
他们的研究提供了对APL发病机制的新证据以及针对APL的潜在治疗选择。
文章标题:
TRIB3 Promotes APL Progression through
Stabilization of the Oncoprotein PML-RARa and
Inhibition of p53-Mediated Senescence
- 浏览 6755 次

