







(1) 目的基因片断扩增
1.引物设计
利用DNAMAN等软件设计合适的引物, 并根据目的基因内切酶位点分析结果,分别在正、反向引物的5’端加上相应酶切位点,并分别加上内切酶保护碱基。
2.按照如下体系进行PCR扩增,测试引物
ddH2O | 16.0 ul |
10×PCRbuffer(with 15uM Mg2+) | 2.0ul |
dNTP(10mM) | 0.4ul |
primer(F/R;10uM) | 0.4ul |
Template DNA(100ng/ul) | 1.0ul |
Taq(5U/ul) | 0.2ul |

扩增产物用1%琼脂糖凝胶电泳检测。
3. 按照如下体系高保真扩增启动子DNA
50ul体系:
ddH2O | 33.5 ul |
KOD-plus 10×PCR buffer | 5.0ul |
MgSO4(25mM) | 2.0ul |
dNTP(2mM) | 5.0ul |
primer(F/R;10uM) | 1.5ul |
Template DNA | 2.0ul |
KOD-plus(1U/ul) | 1.0ul |
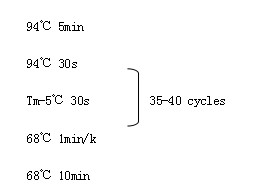
扩增产物用1%琼脂糖凝胶电泳检测。
(2)目的基因片断回收(离心柱型琼脂糖凝胶DNA回收试剂盒)
1.配制1%琼脂糖凝胶,然后将PCR产物全部点入胶孔,进行琼脂糖凝胶电泳。
2.在紫外灯下切下含有目的DNA的琼脂糖凝胶,用纸巾吸尽凝胶表面液体。
3.用电子天平称量凝胶的重量,以1 mg=100ul换算凝胶体积。
4.加入3倍胶体积溶胶液,50℃水浴放置10分钟,其间不断上下翻转(温和的)至胶完全溶解,放置至室温(这步很重要,因为高温不利于DNA吸附)。
5.将已经溶解的溶液加至吸附柱,并将吸附柱放入收集管中,8000rpm离心30s;将收集管中的液体重复加至吸附柱,8000rpm离心30s,将收集管中的废液倒掉。
6.向吸附柱中加入700ul漂洗液(确认已加入乙醇),12000rpm离心30s,将收集管中的废液倒掉。向吸附柱中加入500ul漂洗液重复操作一次。
7.12000rpm 离心2min ,尽量去除漂洗液,在37 ℃培养箱中晾5分钟。
8.吸附柱放入一个干净的离心管,加入30ul 65~ 70℃预热的ddH2O,室温下静置2min,13000rpm离心1min。将离心得到的溶液加入吸附柱中重复操作一次。
9.1%琼脂糖凝胶电泳检测并根据marker的上样量估计回收产物的浓度。
(3)目的片断连入pGEM-T easy 载体(或直接进入(4)酶切)
① PCR扩增产物末端加腺嘌呤核苷酸(A)
1.取用KOD-plus聚合酶扩增的PCR产物1ul,加至10ul Taq PCR 体系。
2.70℃温育15到30分钟
② PCR产物连接入pGEM-T easy载体
连接体系
2×Rapid Ligation buffer | 5ul |
Vector (50ng/ul) | 1ul |
PCR product | Xul |
T4 DNA Ligase (3weiss units/ul) | 1ul |
X的值为:
载体质量(ng) × 插入片断大小(Kb)×[ 插入片断:载体的摩尔比(3:1)]
载体的大小(Kb)
最后补水至10ul
同时设立对照组:
2×Rapid Ligation buffer | 5ul |
Vector (50ng/ul) | 1ul |
Control insert DNA (4ng/ul) | 2 ul |
T4 DNA Ligase (3weiss units/ul) | 1 ul |
ddH2O | 1ul |
(4)对载体和回收得到的目的基因片断进行酶切
1. 在Fermentas网站上(http://www.fermentas.com/doubledigest/index.html)确认所用内切 酶双切体系。一般酶用量为10U/ug;酶的体积不能超过酶切体系的十分之一;体积允许的情况下用30ul酶切体系。以下以EcoRⅠ- VspⅠ双切为例。
2. 载体酶切体系
ddH2O | 13.0 ul |
10×tango buffer | 4.0 ul |
Vector(1ug/ul) | 1.0 ul |
EcoRⅠ (10U/ul) | 1.0 ul |
VspⅠ (10U/ul) | 1 .0ul |
同时设立两个对照:
ddH2O | 14.0 ul |
10×tango buffer | 4 .0ul |
Vector (1ug/ul) | 1.0 ul |
EcoRⅠ (10U/ul) | 1.0 ul |
VspⅠ (10U/ul) | --- |
ddH2O | 14.0 ul |
10×tango buffer | 4.0 ul |
Vector (1ug/ul) | 1 .0ul |
EcoRⅠ (10U/ul) | --- |
VspⅠ (10U/ul) | --- |
370C酶切3个小时;1%琼脂糖凝胶电泳进行鉴定。
3. 目的基因片断酶切体系:
ddH2O | 8 .0ul |
10×tango buffer | 10.0 ul |
hCN promoter (25ng/ul) | 30.0 ul |
EcoRⅠ (10U/ul) | 1.0 ul |
VspⅠ (10U/ul) | 1.0 ul |
370C酶切2个小时; 1%琼脂糖凝胶电泳进行鉴定。
4.载体和目的基因片断回收
1.方法如上述。
2.回收电泳是对insert和vector进行定量,确定其相对质量比例。根据相对分子量换算为两者摩尔比。
(5) 回收产物酶切进行连接反应
连接反应体系:
10× ligase reaction buffer | 2ul (室温溶至无沉淀) |
Vector | 3ul |
Insert | 14ul |
T4 ligase | 1ul |
ddH2O | 添加到20ul |
同时设立自连对照:
10× ligase reaction buffer | 2ul (室温溶至无沉淀) |
Vector | 3ul |
T4 ligase | 1ul |
ddH2O | 14ul |
23℃连接1小时,或16℃连接过夜。
注:对于大小在500bp -1500bp的片段,可以不定量按照上述标准体系连接。连接体系总的DNA量在50-250ng。
(6) 连接产物转化感受态大肠杆菌(E.coli)
1.从-70℃冰箱中取出感受态E.coli(100ul一管),立即放入冰中。
2.5分钟之后感受态E.coli开始溶化时分装为50ul/tube,加入连接产物10ul,冰上放置20min。
3.42℃水浴60-90s;之后冰上放置2分钟(这一步注意动作尽量轻)。
4.加入新鲜的无选择性LB培养基450ul,37℃水浴60分钟。
5.4000rpm离心5min,留100ul上清
6.取Kan/Amp抗性LB平板,加入100ul转化后的感受态大肠杆菌(E.coli)涂板,37℃培养箱培养14-16小时。
7.第二天观察结果,从转化的细菌平板中挑取中等大小,饱满而且没有和其他克隆相接触的单克隆,接种到3ml 卡那霉素选择性LB培养基中。置于37ºC恒温摇床培养14~16个小时。准备质粒的小量抽提。
(7) 阳性克隆鉴定
① 菌落PCR法
1. 配Taq PCR体系母液,按每个平板挑8管,20ul/管计算。
2. 取一PCR排管,每孔加入ddH2O 10ul,做好相应编号;另外准备相应抗性的干净平板一块,做好编号标记。
3.用小枪头从转化的细菌平板中挑取中等大小,饱满且没有和其他克隆相接触的单克隆,在平板上相应编号处轻点一下接种,然后放入相应编号的ddH2O管中。
4.接种平板放入370C培养;取2ul菌悬液进行PCR,剩余8ul菌悬液保存在40C
5.鉴定为阳性的克隆,将8ul菌悬液接种到3mlKan/Amp选择性LB培养基中。置于37ºC恒温摇床培养14~16个小时。若时间间隔过长未摇出菌,可第二天用保种平板接种摇菌。
5.送测序。
② 快速抽提质粒法鉴定
1.从转化的细菌平板中挑取中等大小,饱满而且没有和其他克隆相接触的单克隆,接种到3ml Kan/Amp选择性LB培养基中。置于37ºC恒温摇床培养8小时以上。
2.取30ul菌液(菌较少时可离心后重悬至30ul)加入等体积酚/氯仿,Vortex数次。
3.加入5ul Loading Buffer。
4.12000rpm离心10min(40C)
5.取上清电泳,同阳性Vector比较大小。
5.大小正确的送测序或进一步酶切鉴定。
③ 抽提质粒酶切鉴定
(1)质粒的小量抽提(采用QIANGEN质粒纯化试剂盒进行质粒的小量抽提)
1.取1.5 ml菌液,14000 rpm离心2分钟,然后弃去上清。
2.悬浮:加入P1溶液200 ul,然后使沉淀的菌体悬浮。
3.裂解:加入P2溶液200 ul,然后颠倒4~6次。
4.中和:加入P3溶液200 ul,然后颠倒4~6次。
5.12000 rpm冷冻离心10分钟,然后小心吸取上清液于另一洁净离心管内。
6.加入420 ul异丙醇,混匀,12000 rpm冷冻离心15分钟,然后弃去上清。
7.加入70%乙醇200 ul,冷冻离心5分钟。
8.取出晾干,大约5分钟左右。
9.加入20 ul TE缓冲溶液,反复吹吸50次左右,使质粒DNA溶于TE缓冲溶液之中。取少量样品进行琼脂糖凝胶电泳。
(2)小量抽提质粒内切酶酶切鉴定
选取插入片段上和载体上都有的酶切位点,取相应量质粒进行酶切,看是否符合预计片段大小。
(8)质粒的大量抽提 (采用QIAGEN质粒纯化试剂盒进行质粒的大量抽提)
1.从转化的细菌平板中挑取中等大小,饱满而且没有和其他克隆相接触的单克隆,接种到3ml Kan选择性LB培养基中。37ºC恒温摇床培养14~16个小时。
2.从小量培养物中取1 ml用作小量抽提,然后作酶切鉴定,选取阳性克隆的培养物,以1:500的比例接种到200 ml选择性LB培养基中。37ºC恒温摇床培养14~16个小时。
3.从大量培养物中取700 ul菌液,再加入300ul甘油冻存。
4.把200 ml菌液倒入250 ml离心管中,4ºC,8500rpm离心15分钟。
5.弃去上清,然后倒扣于吸水纸上片刻。然后加入5 ml 4ºC预冷的细胞重悬液buffer P1(确认已经加了Rnase),反复吹打沉淀至没有悬浮的菌块为止。然后将悬浮液转移到50 ml的离心管中。
6.加入5 ml细胞裂解液buffer P2,彻底且轻柔地颠倒4~6次,在室温下孵育不要超过5分钟(从加入buffer P2开始计时,时间过长会使质粒DNA断裂)。注意:样品不要涡旋,否则会有基因组污染。buffer P2用完后也要及时地盖好盖子,以免被空气中的二氧化碳酸化。
7.加入5 ml 4ºC预冷的pH值调节液buffer P3,及时且轻柔地颠倒混匀4~6次,在冰上孵育15分钟。然后再混匀一次样品。4ºC,17500rpm,冷冻离心30分钟。如果上清不够清需再离心1分钟。
8.用4 ml buffer QBT来平衡层析柱。让液体自由留下。
9.然后步骤7中的上清小心的吸到层析柱中,让液体自由流下。
10.用5 ml buffer QC清洗层析柱一次,然后再加5ml buffer QC清洗层析柱。
11.用5 ml buffer QF清洗层析柱洗脱DNA,用一支50 ml离心管收集。
12.在两管洗脱液中各加入0.7倍(3.5 ml)体积的室温异丙醇沉淀DNA,并在沉淀会出现的位置作标记。离心前一定要涡旋。然后4ºC,15000g,离心30分钟。取出时保持离心管平行移动,小心倒去上清。
13.用2 ml室温的70%乙醇清洗沉淀。然后4ºC,15000g,离心10分钟。取出时保持离心管平行移动,小心倒去上清。
14.在空气中干燥沉淀5~10分钟,用400 ul TE缓冲溶液溶解沉淀。
15.测定质粒DNA的260/280值和浓度。
- 浏览 9851 次

